
Getting acquainted with different regression models using scientific data from different sources
- 41 minsSubset selection, regularization
1, Forward stepwise selection
- (-), Download data from http://science.sciencemag.org/content/early/2018/01/17/science.aar3247 (has been a previous hw)
- (-), Load protein levels, and convert them to numerical values if necessary (has been a previous hw)
- (-), Inspect missing values, and drop patients with missing values in protein levels (has been a previous hw)
- (-), Load cancer labels (has been a previous hw)
%pylab inline
import pandas as pd
from sklearn import metrics as ms
from sklearn.preprocessing import normalize
from sklearn.model_selection import train_test_split
import statsmodels.tools as smt
Populating the interactive namespace from numpy and matplotlib
# modified excel file - header names deleted
xls = pd.ExcelFile('aar3247_Cohen_SM_Tables-S1-S11.xlsx')
# protein levels
protein_levels = pd.read_excel(xls, 'Table S6')
cancerTypes = pd.read_excel(xls, 'Table S11')
tumorTypeAndProteinLevels = np.array(['Tumor type','AFP (pg/ml)',
'Angiopoietin-2 (pg/ml)', 'AXL (pg/ml)', 'CA-125 (U/ml)',
'CA 15-3 (U/ml)', 'CA19-9 (U/ml)', 'CD44 (ng/ml)','CEA (pg/ml)',
'CYFRA 21-1 (pg/ml)', 'DKK1 (ng/ml)', 'Endoglin (pg/ml)',
'FGF2 (pg/ml)', 'Follistatin (pg/ml)', 'Galectin-3 (ng/ml)',
'G-CSF (pg/ml)', 'GDF15 (ng/ml)', 'HE4 (pg/ml)', 'HGF (pg/ml)',
'IL-6 (pg/ml)', 'IL-8 (pg/ml)', 'Kallikrein-6 (pg/ml)',
'Leptin (pg/ml)', 'Mesothelin (ng/ml)', 'Midkine (pg/ml)',
'Myeloperoxidase (ng/ml)', 'NSE (ng/ml)', 'OPG (ng/ml)',
'OPN (pg/ml)', 'PAR (pg/ml)', 'Prolactin (pg/ml)',
'sEGFR (pg/ml)', 'sFas (pg/ml)', 'SHBG (nM)',
'sHER2/sEGFR2/sErbB2 (pg/ml)', 'sPECAM-1 (pg/ml)',
'TGFa (pg/ml)', 'Thrombospondin-2 (pg/ml)', 'TIMP-1 (pg/ml)',
'TIMP-2 (pg/ml)'])
# Remove * and ** values
plvls = protein_levels[tumorTypeAndProteinLevels].applymap(
lambda x: float(x[2:]) if type(x)==str and x[0:2]=="**" else x)
plvls = plvls.applymap(
lambda x: float(x[1:]) if type(x)==str and x[0:1]=="*" else x)
# Drop rows containing NaN or missing value
plvls = plvls.dropna(axis='rows')
# Extract cancer types for all the corresponding protein levels
cancer_types = np.array(list(map(lambda x: 1 if x=="Normal" else 0,
plvls['Tumor type'].values)))
plvls = plvls[tumorTypeAndProteinLevels[1:]]
# Normalize
plvls = pd.DataFrame(normalize(plvls, axis=1),
columns=tumorTypeAndProteinLevels[1:])
- (A), Perform forward iterative subset selection using statsmodels logistic regression when predicting the cancer/normal binary labels using protein levels as inputs. Use the log-likelihood values to select the best additional variable in each iteration.
from statsmodels.discrete.discrete_model import LogitResults, Logit
def iteratively_select_variables(data, data_labels, max_variables=3):
max_variables = max_variables if data.columns.size > 3 else data.columns.size//2
# Labels
labels = data.columns
# Best labels
best_labels = []
best_log_losses = []
best_aics = []
best_bics = []
for var in range(max_variables):
log_losses = []
aics = []
bics = []
for label in labels:
# Logistic model on binary cancer -> healthy or not
model = Logit(data_labels, smt.add_constant(data[np.append(best_labels, label)]))
# basinhopping seems to be the best option
result = model.fit(method='basinhopping', full_output=True, disp=False)
log_losses.append(-model.loglike(result.params))
aics.append(result.aic)
bics.append(result.bic)
# Find the min loss fit
min_loss_ind = np.argmin(log_losses)
# Update the output values
best_log_losses.append(log_losses[min_loss_ind])
best_aics.append(aics[min_loss_ind])
best_bics.append(bics[min_loss_ind])
# Update best labels and labels
best_labels = np.append(best_labels, labels[min_loss_ind])
labels = np.delete(labels, [min_loss_ind])
return best_labels, best_log_losses, best_aics, best_bics
labels, losses, aics, bics = iteratively_select_variables(plvls, cancer_types, max_variables=6)
2, Subset selection
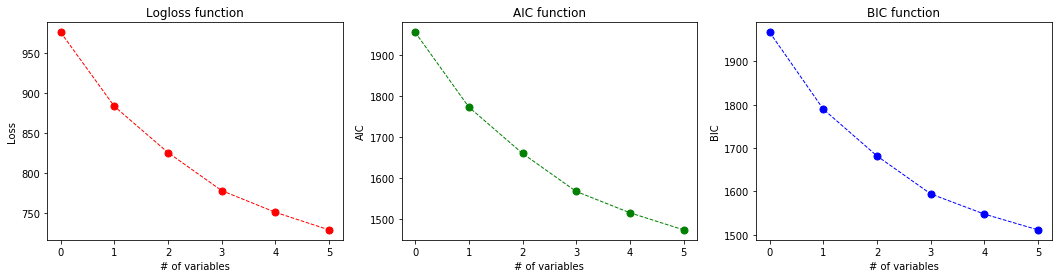
- (A), Plot the AIC and the BIC criterion values depending on the number of variables during forward subset selection. Interpret the result based on the formulas for the 2 criteria!
import itertools
def findsubsets(S,m):
return set(itertools.combinations(S, m))
def plot_output_curves(labels, losses, aics, bics):
plt.figure(figsize=(18, 4))
# Loss function
plt.subplot("131")
plt.plot(range(len(labels)), losses, 'ro--', linewidth=1, markersize=7)
plt.xlabel('# of variables')
plt.ylabel('Loss')
plt.xticks(range(len(labels)))
plt.title("Logloss function")
# AIC function
plt.subplot("132")
plt.plot(range(len(labels)), aics, 'go--', linewidth=1, markersize=7)
plt.xlabel('# of variables')
plt.ylabel('AIC')
plt.xticks(range(len(labels)))
plt.title("AIC function")
# BIC function
plt.subplot("133")
plt.plot(range(len(labels)), bics, 'bo--', linewidth=1, markersize=7)
plt.xlabel('# of variables')
plt.ylabel('BIC')
plt.xticks(range(len(labels)))
plt.title("BIC function")
plot_output_curves(labels, losses, aics, bics)
def plot_aic_bic_difference(aics, bics):
plt.figure(figsize=(8, 4))
# AIC function
plt.plot(range(len(labels)), aics, 'go--', linewidth=1, markersize=7, label='AIC')
plt.xlabel('# of variables')
plt.xticks(range(1, len(labels)+1))
# BIC function
plt.plot(range(len(labels)), bics, 'bo--', linewidth=1, markersize=7, label='BIC')
plt.xlabel('# of variables')
plt.ylabel('AIC/BIC values')
plt.title("AIC/BIC functions")
plt.legend()
plt.show()
plot_aic_bic_difference(aics, bics)
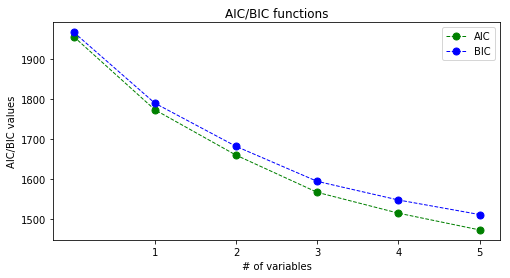
Tha BIC curve is constantly above the AIC curve but the trend seems to be the same. It is due to the fact that the calculation of the two constants is almost the same, but BIC is proportional to log(n)/n while AIC is to 2/n thus AIC is decreasing faster with the increase in n.
- (B), Plot the AUC from a 5-fold cross validation (use sklearn KFold with random_state=0), depending on the number of variables during forward model selection. What do you see compared to the curves from AIC, and BIC?
def iteratively_select_variables_with_cross_validation(data, data_labels, max_variables=3):
max_variables = max_variables if data.columns.size > 3 else data.columns.size//2
# Labels
labels = data.columns
# Best labels
best_labels = []
best_log_losses = []
best_aics = []
best_bics = []
best_aucs = []
for var in range(max_variables):
log_losses = []
aics = []
bics = []
aucs = []
for label in labels:
cross_losses = []
cross_aics = []
cross_bics = []
cross_aucs = []
# 5 fold cross validate
for k in range(5):
X_train, X_test, y_train, y_test = train_test_split(
smt.add_constant(data[np.append(best_labels, label)]), cancer_types,
test_size=0.20, random_state=0)
# Logistic model on binary cancer -> healthy or not
model = Logit(y_train, X_train)
# basinhopping seems to be the best option
result = model.fit(method='basinhopping', full_output=True, disp=False)
y_pred = result.predict(X_test)
fpr, tpr, _ = ms.roc_curve(y_test, y_pred)
cross_losses.append(-model.loglike(result.params))
cross_aics.append(result.aic)
cross_bics.append(result.bic)
cross_aucs.append(ms.auc(fpr, tpr))
log_losses.append(np.mean(cross_losses))
aics.append(np.mean(cross_aics))
bics.append(np.mean(cross_bics))
aucs.append(np.mean(cross_aucs))
# Find the min loss fit
min_loss_ind = np.argmin(log_losses)
# Update the output values
best_log_losses.append(log_losses[min_loss_ind])
best_aics.append(aics[min_loss_ind])
best_bics.append(bics[min_loss_ind])
best_aucs.append(aucs[min_loss_ind])
# Update best labels and labels
best_labels = np.append(best_labels, labels[min_loss_ind])
labels = np.delete(labels, [min_loss_ind])
return best_labels, best_log_losses, best_aics, best_bics, best_aucs
labels, losses, aics, bics, aucs = iteratively_select_variables_with_cross_validation(plvls, cancer_types)
def plot_aic_bic_auc_curves(labels, aics, bics, aucs):
plt.figure(figsize=(16, 4))
# AIC function
plt.subplot("131")
plt.plot(range(len(labels)), aics, 'ro--', linewidth=1, markersize=7)
plt.xlabel('# of variables')
plt.ylabel('AIC')
plt.xticks(range(len(labels)))
plt.title("AIC function")
# AIC function
plt.subplot("132")
plt.plot(range(len(labels)), bics, 'go--', linewidth=1, markersize=7)
plt.xlabel('# of variables')
plt.ylabel('BIC')
plt.xticks(range(len(labels)))
plt.title("BIC function")
# BIC function
plt.subplot("133")
plt.plot(range(len(labels)), aucs, 'bo--', linewidth=1, markersize=7)
plt.xlabel('# of variables')
plt.ylabel('AUC')
plt.xticks(range(len(labels)))
plt.title("AUC function")
plot_aic_bic_auc_curves(labels, aics, bics, aucs)
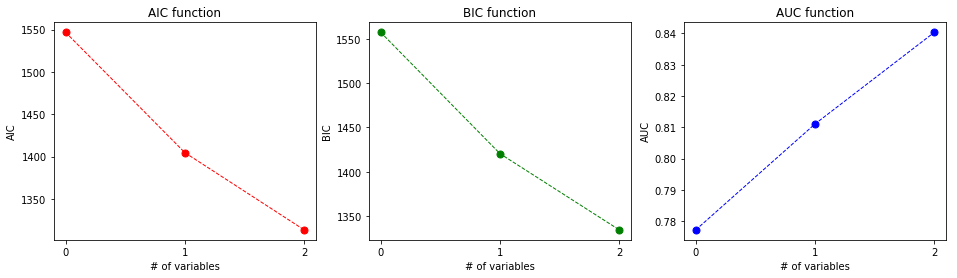
The area under the curve (AUC) gets higher and higher with adding variables. Using only one cross validation for the best labels:
def cross_validate_with_labels(labels, data, data_labels):
cross_losses = []
cross_aics = []
cross_bics = []
cross_aucs = []
# 5 fold cross validate
for k in range(5):
X_train, X_test, y_train, y_test = train_test_split(
smt.add_constant(data[labels]), data_labels,
test_size=0.20, random_state=0)
# Logistic model on binary cancer -> healthy or not
model = Logit(y_train, X_train)
# basinhopping seems to be the best option
result = model.fit(method='basinhopping', full_output=True, disp=False)
y_pred = result.predict(X_test)
fpr, tpr, _ = ms.roc_curve(y_test, y_pred)
cross_losses.append(-model.loglike(result.params))
cross_aics.append(result.aic)
cross_bics.append(result.bic)
cross_aucs.append(ms.auc(fpr, tpr))
return cross_losses, cross_aics, cross_bics, cross_aucs
def plot_cross_validated_data(labels, data, data_labels):
loss, aics, bics, aucs = cross_validate_with_labels(labels, data, data_labels)
plt.figure(figsize=(10, 10))
x = range(5)
# Loss function
plt.subplot("221")
plt.plot(x, loss, 'ro--', linewidth=1, markersize=7)
plt.xlabel('# of variables')
plt.ylabel('Loss')
plt.xticks(x)
plt.title("Loss function")
# AIC function
plt.subplot("222")
plt.plot(x, aics, 'ro--', linewidth=1, markersize=7)
plt.xlabel('# of variables')
plt.ylabel('AIC')
plt.xticks(x)
plt.title("AIC function")
# BIC function
plt.subplot("223")
plt.plot(x, bics, 'go--', linewidth=1, markersize=7)
plt.xlabel('# of variables')
plt.ylabel('BIC')
plt.xticks(x)
plt.title("BIC function")
# AUC function
plt.subplot("224")
plt.plot(x, aucs, 'bo--', linewidth=1, markersize=7)
plt.xlabel('# of variables')
plt.ylabel('AUC')
plt.xticks(x)
plt.title("AUC function")
plot_cross_validated_data(labels, plvls, cancer_types)
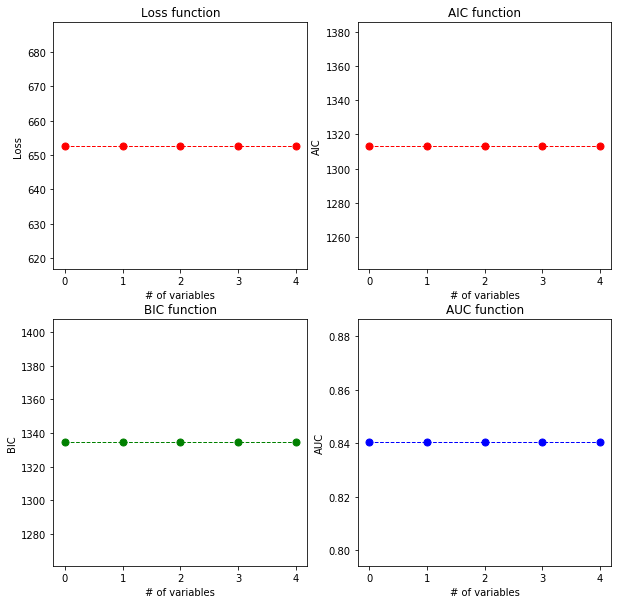
3, Comparison of tools
- (-), Estimate the AUC of a fit with 5-fold cross validation (use sklearn KFold with random_state=0) using the logistic regression from statsmodels using all variables. (has been a previous hw)
- (-), Repeat it with logistic regression fom sklearn. (has been a previous hw)
- (A), Sci-kit learn will be less accurate than statsmodels in this dataset. What are the resons behind the difference?
- (B), Try to fix the problems, and produce a fit witk sklearn which has a 5-fold CV AUC very close (diff<0.0001) to the one with statmodels.
# Cross validate
def cross_validate(plvls, cancer_types, k_fold=5):
logloss = []
accuracy = []
fpr = dict()
tpr = dict()
auc = []
for i in range(k_fold):
# Splitting the randomized data 20%-80%
X_train, X_test, y_train, y_test = train_test_split(smt.add_constant(plvls), cancer_types,
test_size=1./k_fold,
random_state=137*i + i*42, shuffle=True)
# Logistic model on binary cancer -> healthy or not
logistic_model = Logit(y_train, X_train)
# basinhopping seems to be the best option
results = logistic_model.fit(method='basinhopping', full_output=True, disp=False)
# logloss is the negative loglike function
logloss.append(-logistic_model.loglike(results.params))
train_prediction = results.predict(X_test)
pred = train_prediction
truth = y_test
accuracy.append(100.*ms.accuracy_score(truth, np.array(list(map(lambda x: 1 if x > 0.5 else 0, train_prediction)))))
fpr[i], tpr[i], threshold = ms.roc_curve(truth, pred)
auc.append(ms.auc(fpr[i], tpr[i]))
return logloss, accuracy, auc, fpr, tpr
def plot_cross_validated_data(values, k_fold=5):
logloss, accuracy, auc, fpr, tpr = values
print("Mean Accuracy = %.4f%%" % np.mean(accuracy))
if(len(logloss) > 0):
print("\t\t Mean logloss = %.3f" % np.mean(logloss))
plt.figure(figsize=(8.,8.))
lw = 2
cmap = ['r', 'g', 'b', 'c', 'darkorange']
for i in range(k_fold):
plt.plot(fpr[i], tpr[i], color=cmap[i%5],lw=lw, linestyle='--', label="AUC = %.3f " % auc[i])
plt.plot([0, 1], [0, 1], color='navy', lw=lw, linestyle='-', label="Mean AUC = %.3f +/- %.3f" %
(np.mean(auc), np.std(auc)))
plt.xlim([0.0, 1.0])
plt.ylim([0.0, 1.0])
plt.xlabel('False Positive Rate')
plt.ylabel('True Positive Rate')
plt.legend(loc="lower right")
plt.show()
plot_cross_validated_data(cross_validate(plvls.values, cancer_types))
Mean Accuracy = 85.0970%
Mean logloss = 475.912
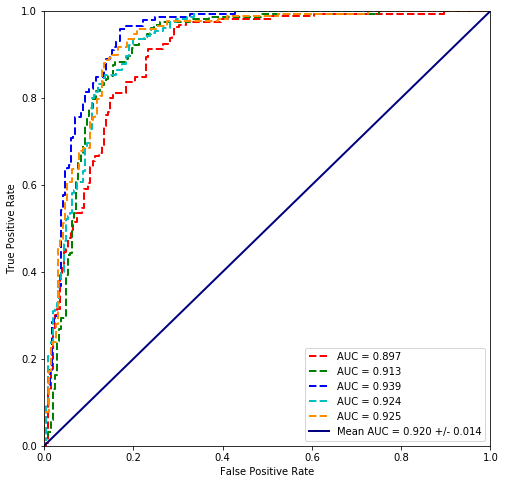
from sklearn.metrics import log_loss
# Cross validate
def scikit_cross_validate(model, plvls, cancer_types, k_fold=5):
logloss = []
accuracy = []
mse = []
fpr = dict()
tpr = dict()
auc = []
for i in range(k_fold):
# Splitting the randomized data 20%-80%
X_train, X_test, y_train, y_test = train_test_split(smt.add_constant(plvls), cancer_types,
test_size=1./k_fold,
random_state=137*i + i*42, shuffle=True)
# Logistic model on binary cancer -> healthy or not
clf = model.fit(X_train, y_train)
train_prediction = clf.predict(X_test)
pred_value = clf.decision_function(X_test)
pred_proba = clf.predict_proba(X_test)
pred = train_prediction
truth = y_test
logloss.append(log_loss(truth, pred_proba))
accuracy.append(100.*ms.accuracy_score(truth, train_prediction))
fpr[i], tpr[i], threshold = ms.roc_curve(truth, pred_value)
auc.append(ms.auc(fpr[i], tpr[i]))
return logloss, accuracy, auc, fpr, tpr
from sklearn.linear_model import LogisticRegression
plot_cross_validated_data(scikit_cross_validate(LogisticRegression(penalty='l1', C=156),
plvls.values, cancer_types))
Mean Accuracy = 85.0970%
Mean logloss = 0.356
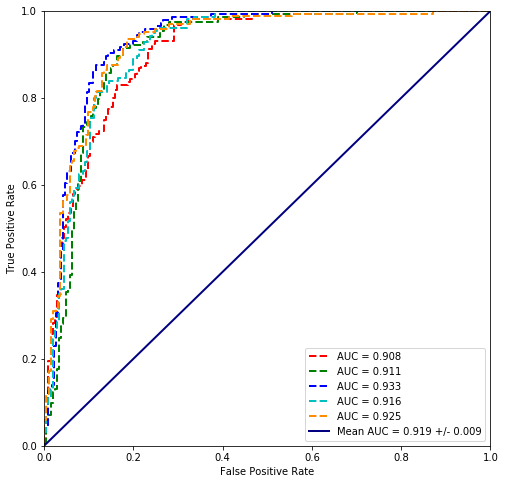
It seems that the scikit regression performs worse which might be do to regularization, which is also possible wih statsmodels. However I am not doing it by using regularized fit via statmodels but trying to disable regularization of the sklearn package. It is not actually possible but setting the C parameter large enough (156) it is possible to calculate them with the same accuracy. reference
4, Large dimensional regression
- Load the prepared data file “methyl.csv” (Which is a subset of the data from the article: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3780611/ )
- (A), Predict the age of people based on methylation values, using linear regression with a 80-20% train-test split (use sklearn train-test split with random_state=0). Plot the scatterplot of true values vs predictions for both the training and the testing set, what do you see?
methyl = pd.read_csv("small_met.csv", sep='\t')
methyl_values = methyl.values[:, 1:methyl.columns.size-1]
ages = methyl['age'].values
from sklearn.linear_model import LinearRegression
X_train, X_test, y_train, y_test = train_test_split(methyl_values, ages, shuffle=True, random_state=0)
clf = LinearRegression().fit(X_train, y_train)
pred_train = clf.predict(X_train)
pred_test = clf.predict(X_test)
def plot_predictions(y_true, y_pred):
plt.figure(figsize=(5, 5))
plt.plot(y_true, y_pred, 'r*', markersize=6, label='Ages')
plt.title("Predicted ages")
plt.xlabel("True ages")
plt.ylabel("Predicted ages")
plt.plot(np.linspace(0, 101, 2), np.linspace(0, 101, 2), 'bo-', markersize=1, linewidth=1, label='Linear')
plt.xlim(0, 100)
plt.ylim(0, 100)
plt.legend()
plt.show()
Predictions for the training data
plot_predictions(y_train, pred_train)
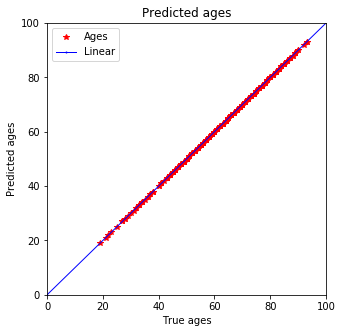
Well, it is not so linear…
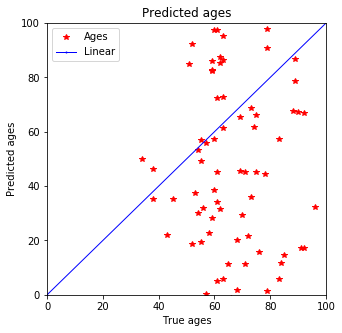
plot_predictions(y_test, pred_test)
- (B), Plot the MSE with 5 fold CV, using Ridge regression, depending on the penalty threshold. Find and optimal penalty value.
- (C), Plot the MSE with 5 fold CV, using lasso regression, depending on the penalty threshold. Find and optimal penalty value. Inspect how many coefficients are 0?
- (D), Use the best model found by the methods above to predict age from methylation with a 80-20% train-test split (use sklearn train-test split with random_state=0). Plot the scatterplot of true values vs predictions for both the training and the testing set.
from sklearn.linear_model import Ridge
from sklearn.metrics import log_loss, mean_squared_error
# Cross validate
def scikit_cross_validate(model, data, data_labels, k_fold=5):
logloss = []
accuracy = []
mse = []
fpr = dict()
tpr = dict()
auc = []
for i in range(k_fold):
# Splitting the randomized data 20%-80%
X_train, X_test, y_train, y_test = train_test_split(smt.add_constant(data), data_labels,
test_size=1./k_fold,
random_state=137*i + i*42, shuffle=True)
# Logistic model on binary cancer -> healthy or not
clf = model.fit(X_train, y_train)
train_prediction = clf.predict(X_test)
pred = train_prediction
truth = y_test
mse.append(mean_squared_error(truth, pred))
return mse, clf.coef_
def plot_mse_in_ridge_alpha_range(model, data, data_labels, ran=np.linspace(0.001, 1, 30), k_fold=5):
i = 1
mses = []
std_mses = []
coefs = []
for val in ran:
mse, coef = scikit_cross_validate(model(alpha=val), data, data_labels, k_fold=k_fold)
mses.append(np.mean(mse))
std_mses.append(np.std(mse))
coefs.append(coef)
plt.figure(figsize=(10,6))
plt.errorbar(ran, mses, yerr=std_mses, fmt='*', elinewidth=1)
plt.xlabel('Alpha')
plt.ylabel('MSE')
plt.xticks(ran[::10])
plt.title("Mean MSE in 5 folds with errors")
plt.show()
np.set_printoptions(precision=3)
plot_mse_in_ridge_alpha_range(Ridge, methyl_values, ages)
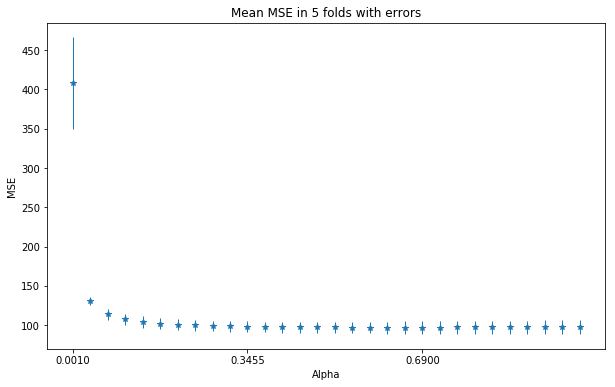
from sklearn.linear_model import Lasso
plot_mse_in_ridge_alpha_range(Lasso, methyl_values, ages)
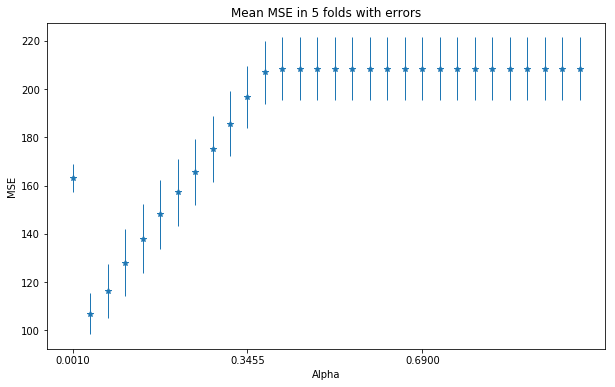
- (D), Use the best model found by the methods above to predict age from methylation with a 80-20% train-test split (use sklearn train-test split with random_state=0). Plot the scatterplot of true values vs predictions for both the training and the testing set.
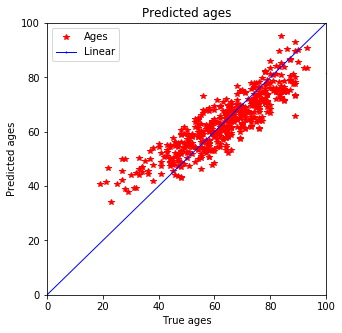
Seeing the examples above the Ridge regression is the candidate for sure. The regression seesm stable from 0.35 thus I’ll set the alpha to be sure to .69
X_train, X_test, y_train, y_test = train_test_split(methyl_values, ages, shuffle=True, random_state=0)
clf = Ridge(alpha=0.69).fit(X_train, y_train)
pred_train = clf.predict(X_train)
pred_test = clf.predict(X_test)
Training prediction
plot_predictions(y_train, pred_train)
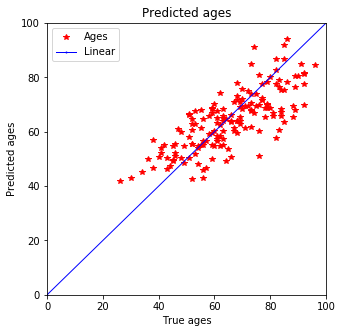
Test prediction - actually seems much better as before
plot_predictions(y_test, pred_test)
5, Shrinkage and coefficient selection
- (A), Show on one plot each coefficient with a line using Ridge regression, depending on the penalty threshold.
- (B), Show on one plot each coefficient with a line using Lasso regression, depending on the penalty threshold.
def plot_coefs(model, data, data_labels, ran=np.linspace(0.1, 4, 35), k_fold=5):
coefs = []
for ind, val in enumerate(ran):
_, coef = scikit_cross_validate(model(alpha=val), data, data_labels, k_fold=k_fold)
coefs.append(coef)
coefs = np.array(coefs).T
plt.figure(figsize=(12, 12))
for ind, line in enumerate(coefs):
plt.plot(ran, line, 'r--', markersize=3, linewidth=1)
plt.xlabel('alhpa')
plt.ylabel('coefficient')
plt.title('Regression coeffiecent distro by alpha parameter')
plt.show()
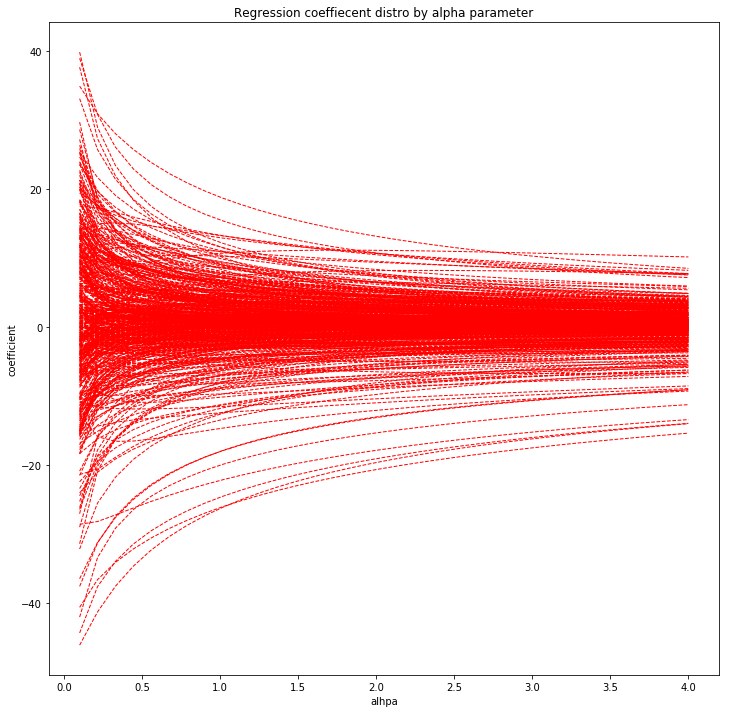
Ridge regression coefficients
plot_coefs(Ridge, methyl_values, ages)
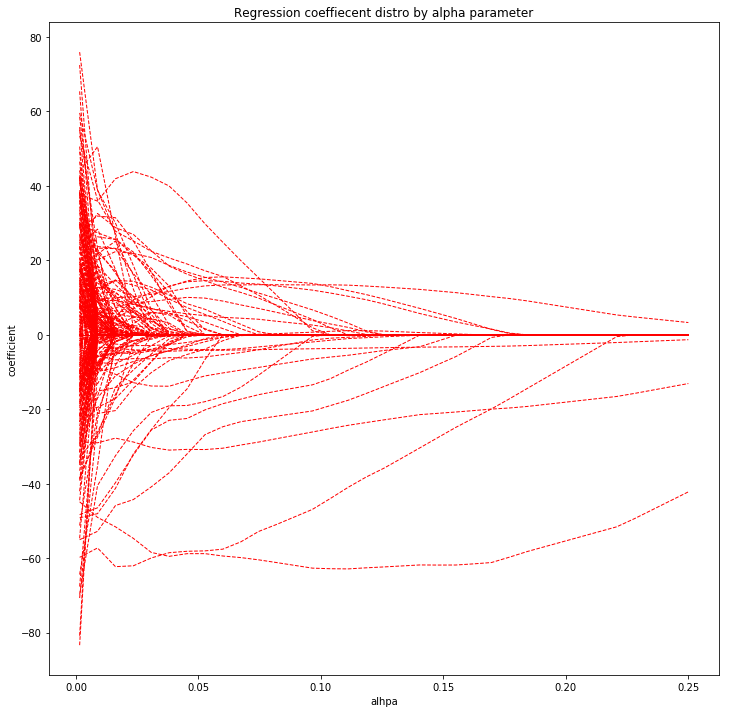
Lasso regression coefficients
plot_coefs(Lasso, methyl_values, ages, ran=np.linspace(0.0015,0.25,35))
- (C), Reuse your code form 1,2 and: Perform forward iterative subset selection with linear regression (use sklearn its much faster). Use the R-squared values to select the best additional variable in each iteration. Plot the AIC and the BIC values and MAE from 5-fold cross validation (use sklearn KFold with random_state=0) depending on the number of variables during forward model selection. Which is the best number of variables? This will take quite some time, at least go up to 150 variables selected.
- (D), Use the best linear regression model found by forward subset selection to predict age from methylation with a 80-20% train-test split (use sklearn train-test split with random_state=0). Plot the scatterplot of true values vs predictions for both the training and the testing set. How does it look compared to 4/(D)?
from statsmodels.regression.linear_model import OLS
def scikit_iteratively_select_variables_with_cross_validation(data, data_labels, max_variables=3):
max_variables = max_variables if data.columns.size > 3 else data.columns.size//2
# Labels
labels = data.columns
# Best labels
best_labels = np.array([])
best_r_squared = []
best_aics = []
best_bics = []
best_maes = []
for var in range(max_variables):
print('Variables: ', var+1, end='\t')
r_squared = []
aics = []
bics = []
maes = []
for label in labels:
cross_r_squared = []
cross_aics = []
cross_bics = []
cross_mae = []
# 5 fold cross validate
for k in range(5):
X_train, X_test, y_train, y_test = train_test_split(
smt.add_constant(data[np.append(best_labels, label)]), data_labels,
test_size=0.20, random_state=0)
clf = OLS(y_train, X_train).fit()
y_pred = clf.predict(X_test)
cross_r_squared.append(clf.rsquared)
cross_aics.append(clf.aic)
cross_bics.append(clf.bic)
cross_mae.append(mean_absolute_error(y_test, y_pred))
r_squared.append(np.mean(cross_r_squared))
aics.append(np.mean(cross_aics))
bics.append(np.mean(cross_bics))
maes.append(np.mean(cross_mae))
# Find the min loss fit
min_ind = np.argmax(r_squared)
# Update the output values
best_r_squared.append(r_squared[min_ind])
best_aics.append(aics[min_ind])
best_bics.append(bics[min_ind])
best_maes.append(maes[min_ind])
# Update best labels and labels
best_labels = np.append(best_labels, labels[min_ind])
labels = np.delete(labels, [min_ind])
return best_labels, best_r_squared, best_aics, best_bics, best_maes
data_columns = methyl.columns[1:][::-1][1:][::-1]
methyl = pd.read_csv("small_met.csv", sep='\t')
methyl_values = methyl.values[:, 1:methyl.columns.size-1]
ages = methyl['age'].values
labels, r_squared, aics, bics, maes = scikit_iteratively_select_variables_with_cross_validation(
methyl[data_columns], ages, max_variables=150)
Variables: 1 Variables: 2 Variables: 3 Variables: 4 Variables: 5 Variables: 6 Variables: 7 Variables: 8 Variables: 9 Variables: 10 Variables: 11 Variables: 12 Variables: 13 Variables: 14 Variables: 15 Variables: 16 Variables: 17 Variables: 18 Variables: 19 Variables: 20 Variables: 21 Variables: 22 Variables: 23 Variables: 24 Variables: 25 Variables: 26 Variables: 27 Variables: 28 Variables: 29 Variables: 30 Variables: 31 Variables: 32 Variables: 33 Variables: 34 Variables: 35 Variables: 36 Variables: 37 Variables: 38 Variables: 39 Variables: 40 Variables: 41 Variables: 42 Variables: 43 Variables: 44 Variables: 45 Variables: 46 Variables: 47 Variables: 48 Variables: 49 Variables: 50 Variables: 51 Variables: 52 Variables: 53 Variables: 54 Variables: 55 Variables: 56 Variables: 57 Variables: 58 Variables: 59 Variables: 60 Variables: 61 Variables: 62 Variables: 63 Variables: 64 Variables: 65 Variables: 66 Variables: 67 Variables: 68 Variables: 69 Variables: 70 Variables: 71 Variables: 72 Variables: 73 Variables: 74 Variables: 75 Variables: 76 Variables: 77 Variables: 78 Variables: 79 Variables: 80 Variables: 81 Variables: 82 Variables: 83 Variables: 84 Variables: 85 Variables: 86 Variables: 87 Variables: 88 Variables: 89 Variables: 90 Variables: 91 Variables: 92 Variables: 93 Variables: 94 Variables: 95 Variables: 96 Variables: 97 Variables: 98 Variables: 99 Variables: 100 Variables: 101 Variables: 102 Variables: 103 Variables: 104 Variables: 105 Variables: 106 Variables: 107 Variables: 108 Variables: 109 Variables: 110 Variables: 111 Variables: 112 Variables: 113 Variables: 114 Variables: 115 Variables: 116 Variables: 117 Variables: 118 Variables: 119 Variables: 120 Variables: 121 Variables: 122 Variables: 123 Variables: 124 Variables: 125 Variables: 126 Variables: 127 Variables: 128 Variables: 129 Variables: 130 Variables: 131 Variables: 132 Variables: 133 Variables: 134 Variables: 135 Variables: 136 Variables: 137 Variables: 138 Variables: 139 Variables: 140 Variables: 141 Variables: 142 Variables: 143 Variables: 144 Variables: 145 Variables: 146 Variables: 147 Variables: 148 Variables: 149 Variables: 150
def plot_r2_aics_bics_maes_data(r2, aics, bics, maes, max_variables):
plt.figure(figsize=(15, 15))
x = range(max_variables)
xx = range(0,max_variables,max_variables//10)
# Loss function
plt.subplot("221")
plt.plot(x, r2, 'ro--', linewidth=1, markersize=7)
plt.xlabel('# of variables')
plt.ylabel('r2')
plt.xticks(xx)
plt.title("r-squared function")
# AIC function
plt.subplot("222")
plt.plot(x, aics, 'ro--', linewidth=1, markersize=7)
plt.xlabel('# of variables')
plt.ylabel('AIC')
plt.xticks(xx)
plt.title("AIC function")
# BIC function
plt.subplot("223")
plt.plot(x, bics, 'go--', linewidth=1, markersize=7)
plt.xlabel('# of variables')
plt.ylabel('BIC')
plt.xticks(xx)
plt.title("BIC function")
# AUC function
plt.subplot("224")
plt.plot(x, maes, 'bo--', linewidth=1, markersize=7)
plt.xlabel('# of variables')
plt.ylabel('MAE')
plt.xticks(xx)
plt.title("MAE function")
plot_r2_aics_bics_maes_data(r_squared, aics, bics, maes, 150)
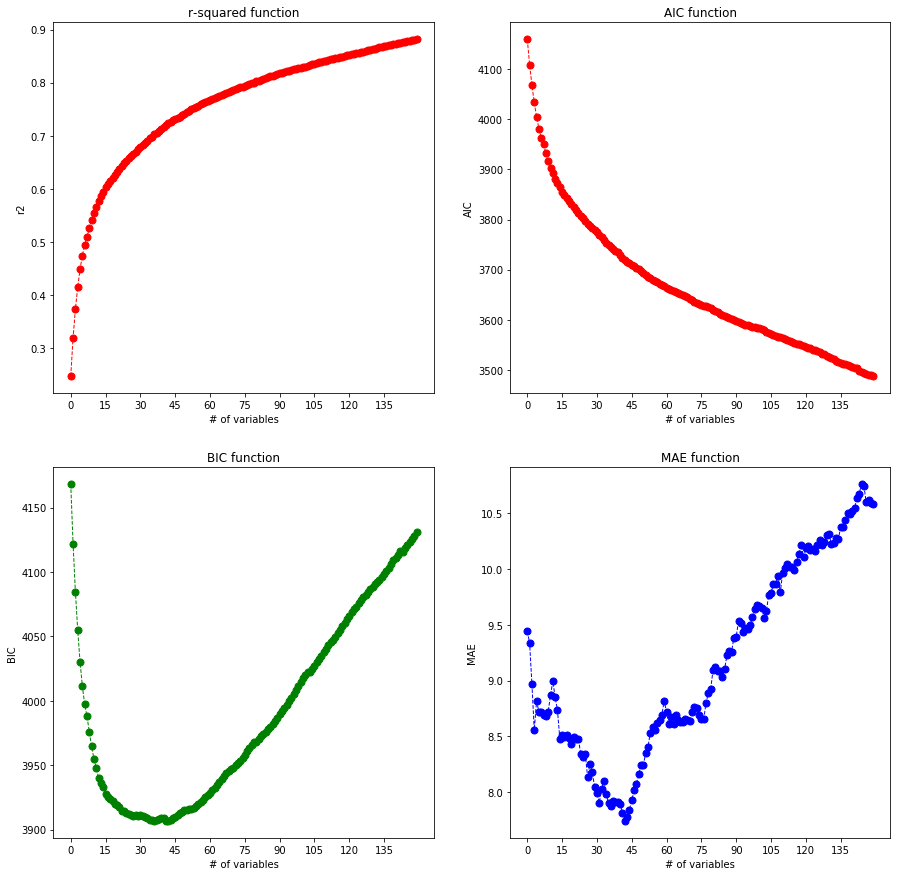
It can be seen that BIC and MAE have their minima at around 42 but AIC is constantly getting smaller, meanwhile r-squared is approaching 1 thus the fit is getting better. Selecting the minimum of the returned BIC and MAE arrays I’m going to take the average of their minimum index and select that as best iterative parameter.
min_bic_ind = np.argmin(bics)
min_mae_ind = np.argmin(maes)
min_ind = max(min_bic_ind, min_mae_ind, (min_bic_ind+min_mae_ind)//2)
min_ind
42
Obviously this MUST be right since it happened to be 42, I should have expected that to be the case all along. Doing the OLS fit with these parameters:
best_labels = labels[:min_ind]
methyl = pd.read_csv("small_met.csv", sep='\t')
methyl_values = methyl[best_labels].values
ages = methyl['age'].values
X_train, X_test, y_train, y_test = train_test_split(
smt.add_constant(methyl_values), ages,
test_size=0.20, random_state=0)
model = OLS(y_train, X_train).fit()
pred_train = model.predict(X_train)
pred_test = model.predict(X_test)
plot_predictions(y_train, pred_train)
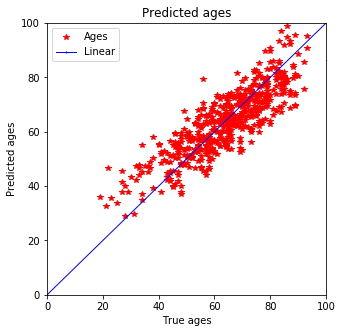
plot_predictions(y_test, pred_test)
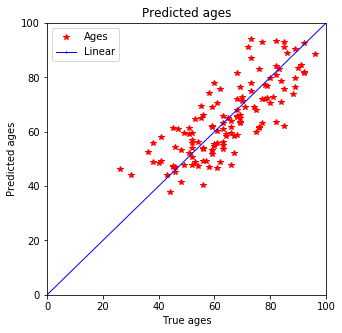
Expecting it qualitatively this prediction seems better than the one with Ridge estimation. Not only does the training data fit better but also the testing data shows smaller discrepancy for younger peoply. Previously a tail was predicted for people below 40 but now that is defeinetly smaller and most of the data is in 3$\sigma$.
@Regards, Alex

Alex Olar
Christian, foodie, physicist, tech enthusiast